StephenMcGowan
asked on
Modifying an existing perl script
Hi,
I'm trying to modify an existing perl script: Speciesmatch.pl
The script currently takes CSV files in the following naming convention: 20130730-p12-A1.csv; i.e. "date, p-number, location.csv"
However, the structure of these files has now changed in that the file names have now been shortened to simply their location (e.g. "A1") followed with a short reference name (e.g. "DOL1").
I've attached 3 example csv data files to this question.
Briefly about the script:
The script (speciesmatch.txt) should process the CSV files one at a time.
For each csv file, it should look at the mass column (first column) and go through the column of scores.
For each score it should take this value and check this mass score against the scores for each animal species contained in SpeciesId.txt. It should record a running tally to see which animal species has the most number of matches to the mass column in the CSV file. The results are recorded in a SpeciesID file where the "top 5 species" are recorded with their number of matches and percentage match.
The script is already set up correctly to do this, although I'm struggling to run it with the altered ".csv" file naming convention (A1-DOL1.csv)
One other modification I'd like to make to this script is to take the top 5 animal species match for a file (say in this case A1)
1. Dolphin 12 matches 100%
2. Cow 5 matches...
3. Snake 2 matches...
etc
Take the very top match-up (in this case Horse)
I would then like the script to open the A1_DOL1.csv file and then write to the very first row:
"Best match: Dolphin"
This would mean that the "mass/intensity/filename/s
Sorry to come across as long winded... but I just thought I'd put as much detail into this question as possible, as I found it quite hard to explain what I'm trying to achieve out of this. Hopefully the script will make sense and will have most of the hard work already done.
Any help with this would be very much appreciated.
Thanks,
Stephen.
Speciesmatch.txt
A1-DOL1.csv
A2-HOR.csv
A3-HIPP.csv
SpeciesId.txt
I'm trying to modify an existing perl script: Speciesmatch.pl
The script currently takes CSV files in the following naming convention: 20130730-p12-A1.csv; i.e. "date, p-number, location.csv"
However, the structure of these files has now changed in that the file names have now been shortened to simply their location (e.g. "A1") followed with a short reference name (e.g. "DOL1").
I've attached 3 example csv data files to this question.
Briefly about the script:
The script (speciesmatch.txt) should process the CSV files one at a time.
For each csv file, it should look at the mass column (first column) and go through the column of scores.
For each score it should take this value and check this mass score against the scores for each animal species contained in SpeciesId.txt. It should record a running tally to see which animal species has the most number of matches to the mass column in the CSV file. The results are recorded in a SpeciesID file where the "top 5 species" are recorded with their number of matches and percentage match.
The script is already set up correctly to do this, although I'm struggling to run it with the altered ".csv" file naming convention (A1-DOL1.csv)
One other modification I'd like to make to this script is to take the top 5 animal species match for a file (say in this case A1)
1. Dolphin 12 matches 100%
2. Cow 5 matches...
3. Snake 2 matches...
etc
Take the very top match-up (in this case Horse)
I would then like the script to open the A1_DOL1.csv file and then write to the very first row:
"Best match: Dolphin"
This would mean that the "mass/intensity/filename/s
Sorry to come across as long winded... but I just thought I'd put as much detail into this question as possible, as I found it quite hard to explain what I'm trying to achieve out of this. Hopefully the script will make sense and will have most of the hard work already done.
Any help with this would be very much appreciated.
Thanks,
Stephen.
Speciesmatch.txt
A1-DOL1.csv
A2-HOR.csv
A3-HIPP.csv
SpeciesId.txt
This should do what you are asking:
#!/usr/bin/perl
use strict;
use warnings;
use Tie::File;
my $len = 0; # hack global because it's simpler
##########################################################################
#Script to identify animal species using monoisotopic peak markers against
#MS data
##########################################################################
# forward slashes in dir name should work
my $dir = 'C:/Users/Stephen/Desktop/20130219_PH12A/relmonopeaklists';
chdir $dir or die "could not cd to $dir: $!";
# create or overwrite SpeciesId
open my $out, '>', 'SpeciesId' or die "could not write SpeciesId: $!";
##########################################################################
#FILE HANDLING
##########################################################################
# get the list of csv files
opendir DIR, '.' or die "could not open dir: $!";
my @files = sort grep m{^[A-P]\d+[-_]\w+\.csv$}, readdir DIR;
# match A-P followed by at least 1 number followed by
## _ or - (description says - but code says _) and any letters/numbers
# alternately, you could simply do m{\.csv$} as trappa suggested but it may
## pick up files you don't want it to
closedir DIR;
####################
#FOR EACH CSV FILE:
####################
foreach my $fil (@files) {
# get wellposition from filename
my ($wellposition) = $fil =~ m{^\d+_\w+_([A-P]\d+)\.csv$};
open my $in, '<', $fil or die "could not open $fil: $!";
# record all masses from the file
my %masses;
while (<$in>) {
chomp;
# skip header line
next if m{mass.*intensity};
my ($mass) = split /,/;
unless ($mass =~ m{^\d+(?:\.\d+)$}) {
warn "mass ($mass) not a recognized number - skipping";
next;
}
$mass = round($mass);
$masses{$mass}++;
}
close $in;
# pass masses hash to subroutine
my $data = analyze(\%masses);
output($wellposition, $data, $fil);
}
close $out;
##########################################################################
#SUB-ROUTINES
##########################################################################
sub round {
my ($num) = @_;
my ($start, $dig) = $num =~ m{^(\d+(?:\.\d)?)(\d)?};
$start += 0.1 if (defined $dig and $dig >= 5);
# XXXXX - XXXXXX
# remove .0 from end of number
# $start =~ s{\.0$}{};
#add .0 to end of number if no decimal
$start .= '.0' unless ($start =~ m{\.\d$});
return $start;
}
# main sub
{ # closure
my $Z='Z';
sub _init {
open my $in, '<', 'SpeciesId3.txt' or die "could not open SpeciesId.txt: $!";
my $spec;
while (<$in>) {
chomp;
next if /^\s*$/; # skip blank lines
if (m{^([A-Z]?)\s*=?\s*(\d+(?:\.\d)?)(?:\s+AND\s+(\d+(?:\.\d)?))?\s*$}) {
# handle letter = lines
push @{$species{$spec}{$1||++$Z}}, $2;
push @{$species{$spec}{$1||$Z}}, $3 if $3;
} else {
# handle species name lines
$spec = $_;
$len = length($spec) if (length($spec) > $len);
}
}
close $in;
}
sub analyze {
my ($masses) = @_;
_init() unless %species;
my %data;
# loop over species entries
SPEC:
foreach my $spec (keys %species) {
# loop over each letter of a species
LTR:
foreach my $ltr (keys %{$species{$spec}}) {
# loop over each mass for a letter
foreach my $mass (@{$species{$spec}{$ltr}}) {
# skip to next letter if it is not found
next LTR unless exists($masses->{$mass});
}
# if we get here, all mass values were found for the species/letter
$data{$spec}{cnt}++;
}
}
# add percentages
foreach my $spec (keys %data) {
$data{$spec}{pct} = round($data{$spec}{cnt} / scalar(keys %{$species{$spec}}) * 100);
}
return \%data;
}
} # end closure
##########################################################################
#RESULTS
##########################################################################
{ # begin closure
my $data;
sub _cust_sort {
if ($data->{$b}{pct} == $data->{$a}{pct}) {
return $data->{$b}{cnt} <=> $data->{$a}{cnt};
}
return $data->{$b}{pct} <=> $data->{$a}{pct};
}
sub output {
my ($wellposition, $data, $fil) = @_;
my @order = sort _cust_sort keys %$data;
print {$out} "Wellposition ($wellposition) Results:\n\n",
"Top 5 Species Identities:\n";
# print out the top 5
for my $i (0..4) {
my $spec = $order[$i];
unless ($spec) {
print "no more matches\n";
last; # exit loop
}
printf {$out} "%d) %-${len}s %d matches %0.1f%%\n", $i+1, $spec, $data->{$spec}{cnt}, $data->{$spec}{pct};
}
if ($order[0]) {
# could be done without Tie::File but I find it easiest for modifying files
tie my @lines, 'Tie::File', $fil or die "could not tie $fil: $!";
unshift @lines, "Best match: $order[0]", '';
untie @lines;
}
}
} # end closureASKER
Hi Wilcoxon,
The script seems to be falling over at the moment, with %species requiring explicit package name:
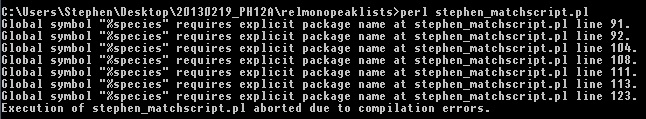
Thanks,
Stephen
The script seems to be falling over at the moment, with %species requiring explicit package name:
Thanks,
Stephen
ASKER
Ok, so I've tried to fix the above problem.
At the beginning of the script I declared %species using
my %species;
The script seems to run through to completion, although I'm receiving a large number of error messages which seem to center around:
uninitialized value in numeric comparison (<=>) at line 137
uninitialized value in numeric eq (==) at line 136
uninitialized value $wellposition in concatenation (.) or string at line 144
(see the attached file)
Checking the SpeciesId results file, it seems that the wellposition ($wellposition) hasn't been written to the file:
i.e.
Wellposition () Results:
Top 5 Species Identities:
1) Mic Oeconomus 1 matches 1.0%
2) Field Mouse 1 matches 1.5%
Wellposition () Results:
where it should read:
Wellposition (A1) Results:
Top 5 Species Identities:
1) Mic Oeconomus 1 matches 1.0%
2) Field Mouse 1 matches 1.5%
Wellposition (A10) Results:
etc etc
errors.jpg
At the beginning of the script I declared %species using
my %species;
#!/usr/bin/perl
use strict;
use warnings;
use Tie::File;
my $len = 0; # hack global because it's simpler
my %species;The script seems to run through to completion, although I'm receiving a large number of error messages which seem to center around:
uninitialized value in numeric comparison (<=>) at line 137
uninitialized value in numeric eq (==) at line 136
uninitialized value $wellposition in concatenation (.) or string at line 144
(see the attached file)
Checking the SpeciesId results file, it seems that the wellposition ($wellposition) hasn't been written to the file:
i.e.
Wellposition () Results:
Top 5 Species Identities:
1) Mic Oeconomus 1 matches 1.0%
2) Field Mouse 1 matches 1.5%
Wellposition () Results:
where it should read:
Wellposition (A1) Results:
Top 5 Species Identities:
1) Mic Oeconomus 1 matches 1.0%
2) Field Mouse 1 matches 1.5%
Wellposition (A10) Results:
etc etc
errors.jpg
I'll try to take a look tomorrow. It's been a busy week.
ASKER CERTIFIED SOLUTION
membership
This solution is only available to members.
To access this solution, you must be a member of Experts Exchange.
ASKER
Excellent! - does exactly as intended.
^ ==> start of line
\d+ ==> one or more numbers
_ ==> an underscore char
\w ==> any single "word" character (a letter, digit or underscore)
_ ==> an underscore char
[A-P] ==> A capital letter between A & P
\d+ ==> one or more numbers
\.csv$ ==> a . followed by csv followed by end of line ( the backslash is so as not to interpret the .)
In order to know exactly how to modify the line, you need to know what are the location naming conventions. Same for the short reference but you could use ^.+\.csv$
This would match any line ending in .csv